

Stryker Corporation has issued a voluntary recall of yet another product used in orthopedic surgeries. In July of 2012, Stryker recalled its Rejuvenate and ABG II modular-neck hip stems used in conjunction with hip replacement surgeries. More recently, Stryker Corporation has acknowledged defects with its ShapeMatch Cutting Guides used to assist in the positioning of total knee replacement components. On April 10, 2013, Stryker issued an urgent medical device recall of the ShapeMatch Cutting Guides. This was classified as a Class I recall by the U.S. Food and Drug Administration, which means that there is a reasonable probability the product can cause serious adverse health consequences or death.
The ShapeMatch Cutting Guides were used with the Triathlon Knee System. The recalled products are single use, disposable cutting guides used as surgical instruments to assist the surgeon in positioning the total knee replacement components and guiding the marking of bone before the surgeon begins cutting. The product was recalled due to a software defect that resulted in wider cutting ranges. Additionally, Stryker determined that a separate software defect resulted in displayed parameters for the depth of resection or the angle of cut not matching the cutting guides produced. It was determined that these defects could result in injuries, including joint instability, fracture, chronic pain, limited mobility of the knee and the need for revision surgery.
Patients who have had knee replacement surgery during which the ShapeMatch Cutting Guides were used and who feel their knee is not functioning properly should contact their orthopedic surgeon. If it is determined that the patient is experiencing joint instability, fracture, chronic pain, or limited mobility in the knee, any of which could necessitate revision surgery, he or she should contact the experienced defective medical device attorneys at Suthers & Harper on our number 912.232.6767, for a free consultation. Suthers & Harper is presently representing numerous individuals in pending lawsuits against the manufacturers of defective hip replacement and knee replacement products, and has the experience and resources necessary to hold the product manufacturers accountable for their defective products.
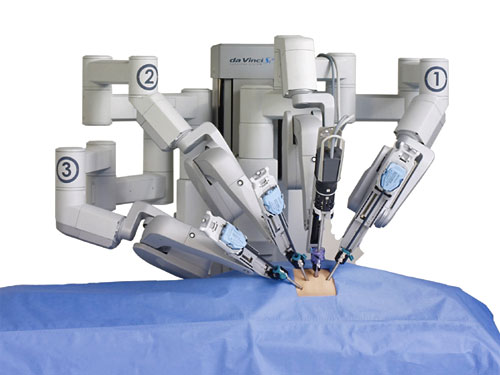 device that has been FDA approved for a range of minimally invasive procedures. During these procedures, the patient is on the operating table while the doctor sits several feet away at the control area of the machine, where he or she can maneuver the robotic arms while watching the procedure through a 3-D viewfinder. Many surgeons feel the robot makes it easier to see and navigate places in the body normally difficult to reach with traditional surgical methods. Doctors advocating the use of the da Vinci also point to the potential benefits for patients, such as small incisions, minimal bleeding, minimal scarring, and speeding post-surgery recovery time. As a result, surgical procedures using the da Vinci robot are becoming increasingly popular in the United States, with close to 400,000 of the procedures performed in the United States in 2012, and more than 1.5 million procedures since Intuitive Surgical received its initial FDA approval of the device.
device that has been FDA approved for a range of minimally invasive procedures. During these procedures, the patient is on the operating table while the doctor sits several feet away at the control area of the machine, where he or she can maneuver the robotic arms while watching the procedure through a 3-D viewfinder. Many surgeons feel the robot makes it easier to see and navigate places in the body normally difficult to reach with traditional surgical methods. Doctors advocating the use of the da Vinci also point to the potential benefits for patients, such as small incisions, minimal bleeding, minimal scarring, and speeding post-surgery recovery time. As a result, surgical procedures using the da Vinci robot are becoming increasingly popular in the United States, with close to 400,000 of the procedures performed in the United States in 2012, and more than 1.5 million procedures since Intuitive Surgical received its initial FDA approval of the device. A number of victims throughout the United States have filed lawsuits against Johnson & Johnson and its subsidiary, McNeil-PPC, after suffering severe liver damage, liver failure and, in some cases, death after taking Tylenol or Acetaminophen. The lawsuits allege that the Defendant manufacturers failed to provide adequate warnings about the risks associated with the side effects of Acetaminophen, which is the active ingredient in Tylenol. The victims suffered serious and life-threatening injuries, even when the medication was taken as directed. Liver damage can lead to prolonged hospitalizations and potentially, the need for a liver transplant. The average cost for a liver transplant in the United States exceeds $500,000.
A number of victims throughout the United States have filed lawsuits against Johnson & Johnson and its subsidiary, McNeil-PPC, after suffering severe liver damage, liver failure and, in some cases, death after taking Tylenol or Acetaminophen. The lawsuits allege that the Defendant manufacturers failed to provide adequate warnings about the risks associated with the side effects of Acetaminophen, which is the active ingredient in Tylenol. The victims suffered serious and life-threatening injuries, even when the medication was taken as directed. Liver damage can lead to prolonged hospitalizations and potentially, the need for a liver transplant. The average cost for a liver transplant in the United States exceeds $500,000.